This website uses cookies to help us give you the best experience when you visit and for certain analytics and marketing activities. By continuing to use this website, you consent to the use of these cookies. Please exit the website if you do not agree to such cookies being used. To find out more about how we use cookies, go to our Privacy Policy.
galactosemia
Learn Plan Connect
As part of the Galactosemia community, we're on this journey together. Let's help one another find our way forward.

What is Galactosemia?
Galactosemia means “too much galactose in the blood.”
Galactose builds up in the blood because the body cannot process this simple sugar.


Take a closer look
The galactose metabolic pathway
There's more to the story about how Galactosemia affects the body.
Step 1
Step 2
Step 3
When everything is working the way it should, galactose is converted by Pathway #1. In this pathway, galactose is turned into galactose-1-phosphate, or Gal-1p by the GALK enzyme.
Because of the inactive or missing GALT enzyme, Gal-1p can't be processed any further. Gal-1p clogs the pathway, so galactose can't go where it's supposed to.
Backed-up galactose overflows into Pathway #2, where it shouldn't go. An enzyme that's not supposed to be involved—Aldose Reductase—changes galactose into toxic galactitol.
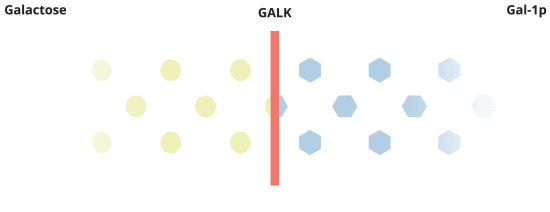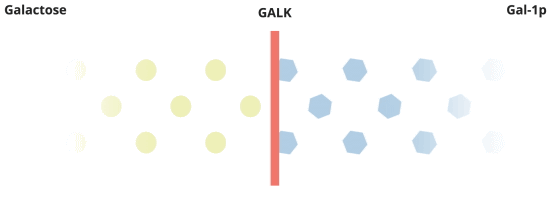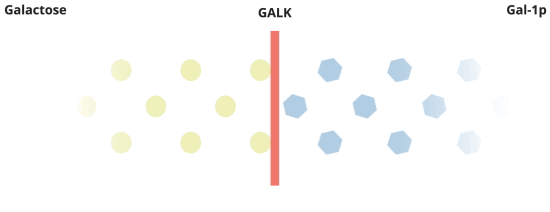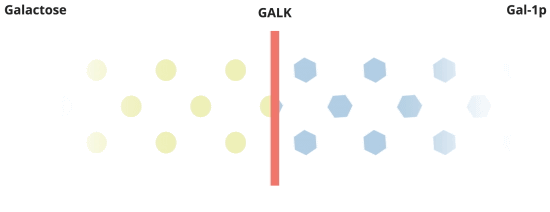


There's more to the story about how Galactosemia affects the body.
Words to Know In Galactosemia
The Four Gs
Galactose
Galactose is a type of sugar found in dairy products and certain foods. It is also produced naturally (endogenously) by the body.
Galactitol
Galactitol is a toxic substance produced in people with Classic Galactosemia. Galactitol has been shown to be responsible for a range of health issues.
Gal-1p
Gal-1p stands for galactose-1-phosphate. Gal-1p is a substance that is typically converted by the body into glucose, which is used for energy. Everybody makes Gal-1p. But in people with Classic Galactosemia, the missing or inactive GALT enzyme leads to an excess build-up of Gal-1p and galactose.
GALT
GALT is an enzyme that helps the body convert Gal-1p into glucose, which is used for energy. Classic Galactosemia is the term used for people who are missing or have an inactive GALT enzyme. GALT is also referred to as galactose-1-phosphate uridylyltransferase.

Cataracts
Speech
issues
issues
Cognitive and developmental delays
Motor issues
Fertility issues
Tremor
Seizures
Making the Connection
The role of toxic galactitol
In Galactosemia, galactose is mistakenly turned into a substance called galactitol. Galactitol is highly toxic, which means it's harmful to the body.
Toxic galactitol can build up in the blood, tissues, and organs, including the brain. There is evidence that toxic galactitol is responsible for a range of health issues that people with Galactosemia may experience.
Rare and Different
Types of Galactosemia
Galactosemia is a rare condition that is diagnosed in approximately 80 newborns in the U.S. each year. In total, it affects 3,000 people in the U.S.
There are several types of Galactosemia. And while each person's experience with Galactosemia will be unique, there are some similar characteristics between each different type.
Classic Galactosemia (CG) (Type I)
This is the most common form of Galactosemia and is caused by an inactive or missing GALT enzyme. The missing GALT enzyme prevents the body from processing galactose and can cause lifelong health complications, some of which can be life threatening at birth.
Galactokinase Deficiency (Type II)
This form of Galactosemia is a result of an inactive or missing GALK enzyme and may have similar outcomes to people with Classic Galactosemia.

Testimonials
Living with Galactosemia
A lot of people say Galactosemia is who you are, but I am just a person with Galactosemia. I don't let Galactosemia define me. Kimberley M. | Patient
Get involved in the Galactosemia community. You'll find help and support there. Heather C. | caregiver
Don't make everything about Galactosemia; the individual is and should be encouraged to be so much more than this condition. Christy J. | caregiver


Stay in the loop and get updates
Connect with Galactosemia Together:
References
- American Liver Foundation. Galactosemia. Accessed September 15, 2020. https://liverfoundation.org/for-patients/about-the-liver/diseases-of-the-liver/galactosemia/
- Berry GT. Classic galactosemia and clinical variant galactosemia. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews®. Seattle (WA): University of Washington; February 4, 2000.
- Coelho AI, Rubio-Gozalbo ME, Vicente JB, Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis. 2017; 40:325-342. doi:10.1007/s10545-017-0029-3
- Data on file. Applied Therapeutics, NY, NY 10017.
- Demirbas D, Coelho AI, Rubio-Gozalbo ME, Berry GT. Hereditary Galactosemia. J Metab. 2018;83:188-196. https://doi.org/10.1016/j.metabol.2018.01.025
- Galactosemia. MedlinePlus US National Library of Medicine. Updated August 18, 2020. Accessed September 18, 2020. https://medlineplus.gov/genetics/condition/galactosemia/
- GALT gene. Medline Plus US National Library of Medicine. Updated August 18, 2020. Accessed October 12, 2020. https://medlineplus.gov/genetics/gene/galt/
- Genomics & Precision Health: Genetics basics. Centers for Disease Control and Prevention. Reviewed March 20, 2020. Accessed October 8, 2020. https://www.cdc.gov/genomics/about/glossary.htm
- Glucose-galactose malabsorption. Medline Plus US National Library of Medicine. Updated August 18, 2020. Accessed October 12, 2020. https://medlineplus.gov/genetics/condition/glucose-galactose-malabsorption/
- Hennermann JB, Schadewalt P, Vetter B, et al. Features and outcome of galactokinase deficiency in children diagnosed by newborn screening. J Inherit Metab Dis. 2011;34:399-407.
- National Organization for Rare Disorders. Galactosemia. Updated 2019. Accessed September 22, 2020. https://rarediseases.org/rare-diseases/galactosemia/
- Ozemir ZA, Tekturk P, Uyguner ZO, Baykan B. Galactosemia and phantom absence seizures. J Pediatr Neurosci. 2014 Sep-Dec; 9(3):253-256. doi:10.4103/1817-1745.147581
- Welling L, Bernstein LE, Berry GT, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017; 40(2):171-176. doi:10.1007/s10545-016-9990-5