This website uses cookies to help us give you the best experience when you visit and for certain analytics and marketing activities. By continuing to use this website, you consent to the use of these cookies. Please exit the website if you do not agree to such cookies being used. To find out more about how we use cookies, go to our Privacy Policy.
Science
Galactosemia
in
the body
What's Really Happening
How Galactosemia Affects the Body
Classic Galactosemia is a rare condition where the body has trouble breaking down a sugar called “galactose.”
People with Galactosemia have a missing or inactive enzyme that is needed to correctly process galactose. Galactose is then converted into a toxic substance called galactitol.
The most common type of Galactosemia that causes health issues is called
“Classic Galactosemia”

Gal-1p
Normally, an enzyme called GALK turns galactose into another substance, galactose-1-phosphate (Gal-1p).

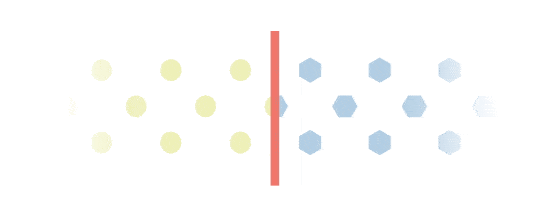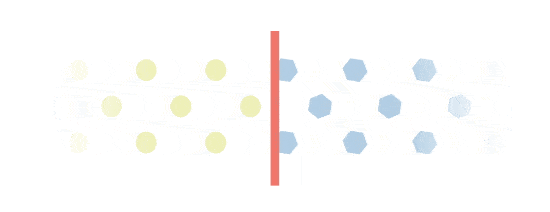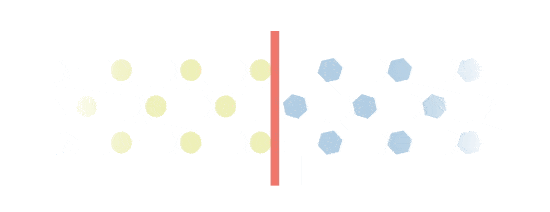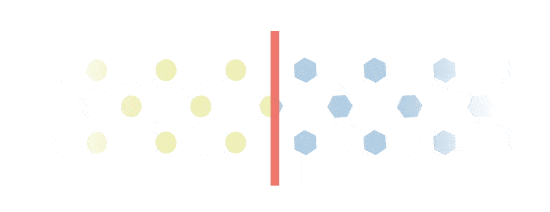
Excess build-up
Everbody makes Gal-1p. However, people with Galactosemia will experience an excess build-up of Gal-1p. This is because they are missing a key enzyme called GALT, which is needed to process Gal-1p. Gal-1p is important in the monitoring of Galactosemia.


Elevated levels
Children and adults with Galactosemia have high levels of Gal-1p in their blood. After an infant has been diagnosed with Galactosemia, their doctor may do a blood test to see how much Gal-1p is in their blood. Once a child with Galactosemia has been on a Galactosemia-friendly diet, their doctor may check their blood again periodically to see how the Gal-1p levels have changed.
Although Gal-1p is monitored, there is no evidence that it directly causes health issues that people with Galactosemia may experience.
Toxic galactitol

Because people with Galactosemia cannot complete the process of metabolizing Gal-1p, excess galactose backs up and is abnormally converted by an enzyme called Aldose Reductase through another pathway that is not usually involved. Then, this enzyme turns galactose into toxic galactitol.


Toxic to the body
Galactitol is harmful to the body. It has been shown that galactitol can build up in the blood, tissues, and organs, including the brain. Using magnetic resonance spectroscopy (MRS) technology, researchers can see build-up of galactitol in the brain.
There is evidence that toxic galactitol is responsible for a range of health issues that people with Galactosemia may experience, including:
Cataracts
Speech issues
Cognitive and developmental delays
Motor issues
Fertility issues
Tremor
Seizures
Only people with Galactosemia make toxic galactitol
This is important to understand because galactitol is harmful to people living with Galactosemia.
Genes
Understanding the genetics of Galactosemia
Galactosemia is a genetic condition, meaning that it is passed down through a child's parents.
When a baby is conceived, it receives genes from each parent, creating thousands of pairs of genes that determine things like the color of their eyes and hair. There is also a gene pair that enables the production of an enzyme that helps our bodies process galactose—this is the GALT gene.
Galactosemia occurs when each of the infant's parents carry one form of the gene that makes a working GALT enzyme and one nonworking gene that does not make the GALT enzyme. When a child inherits a nonworking form of the GALT gene from both parents to form a nonworking GALT gene pair, their GALT enzyme will not work as it should and causes Galactosemia. This kind of genetic inheritance is known as "autosomal recessive" inheritance.


Diagnosis
Blood analyses help healthcare providers diagnose and confirm Galactosemia
GALT mutation analysis
Done to determine the genetic code changes in the GALT gene. Figuring out the type of mutation helps confirm whether the GALT enzyme is missing or to what degree it is functioning.
GALT enzyme activity
Confirms the amount of GALT enzyme activity present in red blood cells—the less GALT enzyme you have, the less you are able to process galactose into energy for the body.

Stay in the loop and get updates
Connect with Galactosemia Together:
References
- Baby's First Test. Classic galactosemia. Updated October 2018. Accessed October 5, 2020. https://www.babysfirsttest.org/newborn-screening/conditions/classic-galactosemia
- Berry GT. Classic galactosemia and clinical variant galactosemia. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews®. Seattle (WA): University of Washington; February 4, 2000.
- Berry GT, et al. In vivo evidence of brain galactitol accumulation in an infant with Galactosemia and encephalopathy. J Ped. 2001;138(2):260-262.
- Coelho AI, Rubio-Gozalbo ME, Vicente JB, Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis. 2017;40:325-342. doi:10.1007/s10545-017-0029-3
- Galactosemia. MedlinePlus US National Library of Medicine. Updated August 18, 2020. Accessed September 18, 2020. https://medlineplus.gov/genetics/condition/galactosemia/
- Genetics basics. Centers for Disease Control and Prevention. Updated March, 2020. Accessed October 1, 2020. https://www.cdc.gov/genomics/about/basics.htm
- Genomics & Precision Health: Genetics basics. Centers for Disease Control and Prevention. Updated March 20, 2020. Accessed October 8, 2020. https://www.cdc.gov/genomics/about/glossary.htm
- Martinelli D et al. Teaching neuroimages: galactitol peak and fatal cerebral edema in classic galactosemia. Am Aca Neurol. 2016;86:e32-e33.
- MedlinePlus Genetics Home Reference: National Library of Medicine (U.S.). Galactosemia. Updated August 18, 2020. https://medlineplus.gov/genetics/condition/galactosemia/
- National Centre for Inherited Metabolic Disorders. Galactosaemia. Updated October 2019. Accessed October 29, 2020. http://metabolic.ie/wp-content/uploads/2019/08/Galactosaemia-Booklet.pdf
- National Organization for Rare Disorders. Galactosemia. Updated 2019. Accessed September 22, 2020. https://rarediseases.org/rare-diseases/galactosemia/
- Otaduy MCG, Leite CG, Lacerda MTC, et al. Proton MR spectroscopy and imaging of a galactosemic patient before and after dietary treatment. AJNR. 2006;27:1-4.
- Rasmussen SA, Daenzer JMI, MacWilliams JA, et al. A galactose-1-phosphate uridylyltransferase-null rat model of classic galactosemia mimics relevant patient outcomes and reveals patient outcomes and reveals tissue-specific and longitudinal differences in galactose metabolism. J Inherit Metab Dis. 2019; 43:518-528. doi:10.1002/jimd.12205
- Rubio-Gozalbo ME, Haskovic M, Bosch AM, et al. The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet J Rare Dis. 2019;14(1):86. doi:10.1186/s13023-019-1047-z
- Welling L, Bernstein LE, Berry GT, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017;40(2):171-176. doi:10.1007/s10545-016-9990-5